Workflow
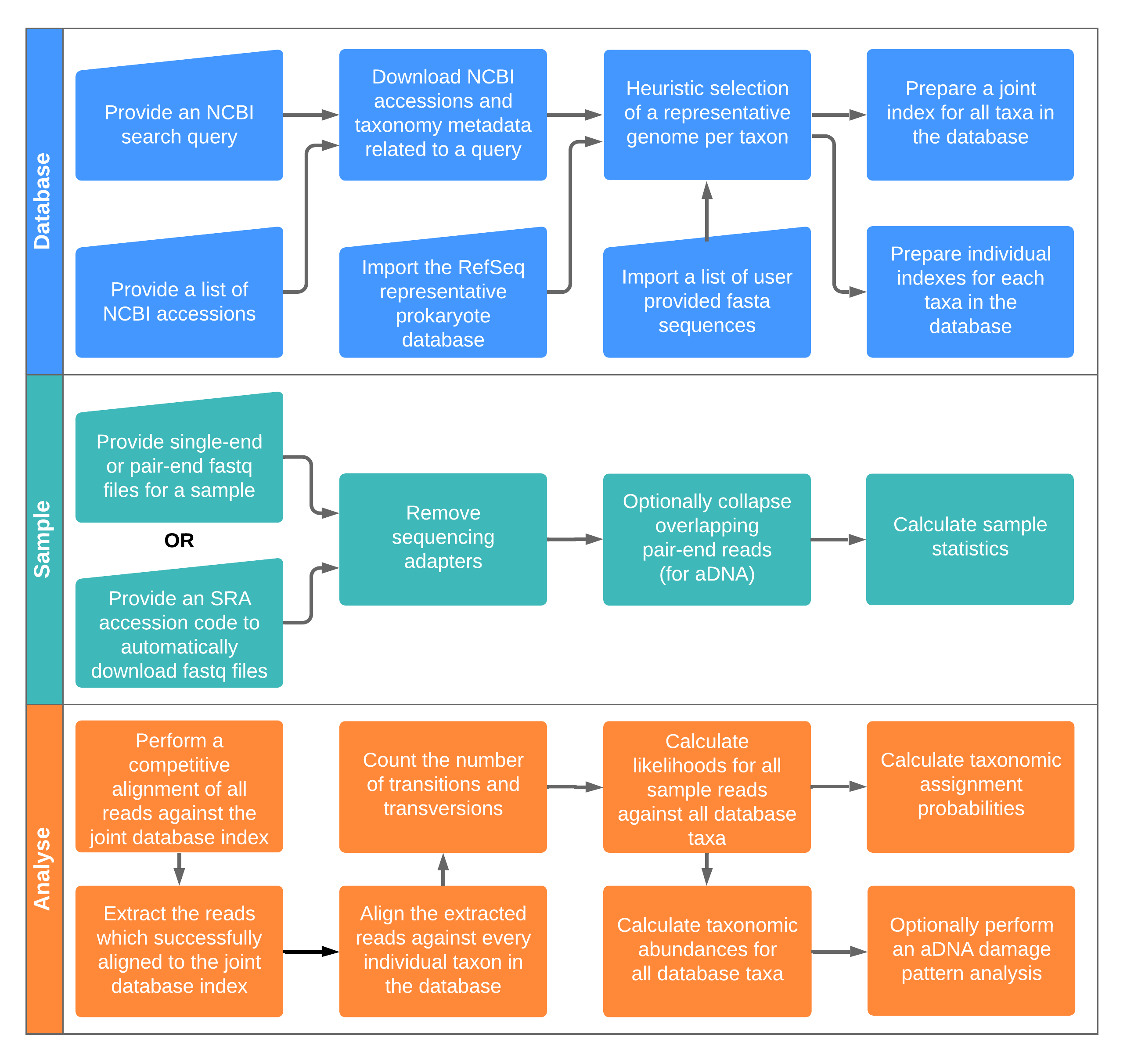
HAYSTAC was designed to be used as a species identifier for either single organism or metagenomic samples. The full execution of the pipeline includes the construction of a database, the processing of a sample file and lastly the analysis of a sample against a specific database. For that purpose three modules have been designed, each of which has its own outputs.
First the haystac database module can be used to construct a database, based on the user’s needs and preferences (custom NCBI query, and/or custom NCBI accessions for each species, and/or prokarytic representative RefSeq, and/or custom sequences). After the user input is collected, the user must provide a path where the outputs of the database can be stored.
The haystac sample module prepares a sample to be analysed. It deals with trimming adapters and collapsing PE reads if needed and it counts the number of reads that are included in the sample file provided by the user.
The haystac analyse module performs the analysis of a sample against a specific database. All its outputs are created under the path that the user specifies with the --output path.